
It is a big challenge in organic synthetic chemistry to construct complex polycyclic compounds efficiently from simple starting materials, especially the ubiquitous hydrocarbons. In the past decade, extensive studies have been directed to the C-H bond activation catalyzed by Cp*Rh(III) or Cp*Co(III) complexes owing to their unique reactivity, selectivity and compatibility with different functional groups.
The research group led by Prof. LI Xingwei from Dalian Institute of Chemical Physics of the Chinese Academy of Sciences made a series of important progress in this area (Angew. Chem. Int. Ed. 2016, 55, 15351 and Angew. Chem. Int. Ed. DOI:10.1002/anie.201704036).
Nitrones are a common 1,3-dipolar reactants in various cycloaddition reactions. The research group firstly applied nitrones as a new versatile directing group in catalytic C-H bond activation in 2013. However, the integration of the C-H bond activation of nitrones with subsequent intramolecular 1,3-dipolar cycloaddition has not been realized.
Recently, they used Cp*Rh(III) to catalyze the coupling of nitrones with diphenylcyclopropenones. The C-H activation step firstly resulted in the acylation and then the activated C=C double bond undergoes intramolecular [3+2] dipolar addition with nitrone to form the bridged bicyclics. The reactions worked well both for N-tert-butyl nitrones with a hindered ortho substituent and N-aryl nitrones. For N-tert-butyl nitrones, the C-H activation occurred at the ortho site of the only aryl ring. For N-aryl nitrones, the C-H activation occurred selectively at the N-aryl ring. Accordingly, the structures of products from these two classes of nitrones were different.
Significantly, when the substituent at the ortho position of N-tert-butyl nitrones is small, the reaction follows C-H activation and insertion into the three-member ring, but the resulting Rh-C(alkenyl)bond was not directly protonated. Instead, it undergoes migratory insertion into the imine moiety followed by ß-carbon elimination and protonolysis to deliver a 1-naphthol. In this case, nitrones acted as a traceless dipolar directing group. These results were published in Angew. Chem. Int. Ed. 2016, 55, 15351.
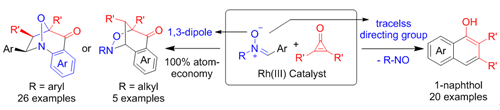
C-H Activation of Nitrones (Image by LI Xingwei, ZHOU Xukai and XIE Fang)
The cyclohexadienone-containing 1,6-enyne moiety has multiple reactive sites, so they have been extensively studied in organic synthesis. However, most reported work only focused on their electrophilicity. The researchers realized a Rh(III) or Co(III) catalyzed divergent coupling between 1,6-enynes and indoles, which combines C-H activation and intramolecular Diels-Alder reaction. Under Rh(III) and Co(III) catalysis, structurally different cyclic products were obtained.
The reactions firstly underwent C-H activation of indole at the 2-position to give a metallacycle. In case of Rh(III) catalysis, a 2,1-insertion of the Rh-C bond into the alkyne occurred, followed by a type-I intramolecular Diels-Alder reaction to form a fused cyclic product. In case of Co(III) catalyst that has a smaller radius, a rare 1,2-insertion of the Co-C bond into the alkyne is followed by a type-II intramolecular Diels-Alder reaction to form a novel [3,3,1]-bicyclic scaffold. These results have been published online (Angew. Chem. Int. Ed. DOI:10.1002/anie.201704036).

Design of Divergent Couplings of Indoles with 1,6-Enynes (Image by LI Xingwei, ZHOU Xukai and XIE Fang)
These works have been financially supported by The National Science Foundation of China and by the Strategic Priority Research Program of the Chinese Academy of Sciences.

86-10-68597521 (day)
86-10-68597289 (night)

86-10-68511095 (day)
86-10-68512458 (night)

cas_en@cas.cn

52 Sanlihe Rd., Xicheng District,
Beijing, China (100864)
Copyright © 2002 - Chinese Academy of Sciences








