
Newsroom





The immune system acts as a critical sentinel of organismal aging, integrating the sensing of physiological states with the execution of defense and clearance functions. Immunosenescence not only reflects systemic functional decline but also serves as a central driver of multiple age-related diseases. However, owing to the high heterogeneity and complexity of the immune system, accurately quantifying its aging status and identifying actionable intervention targets have been major scientific challenges in the field.
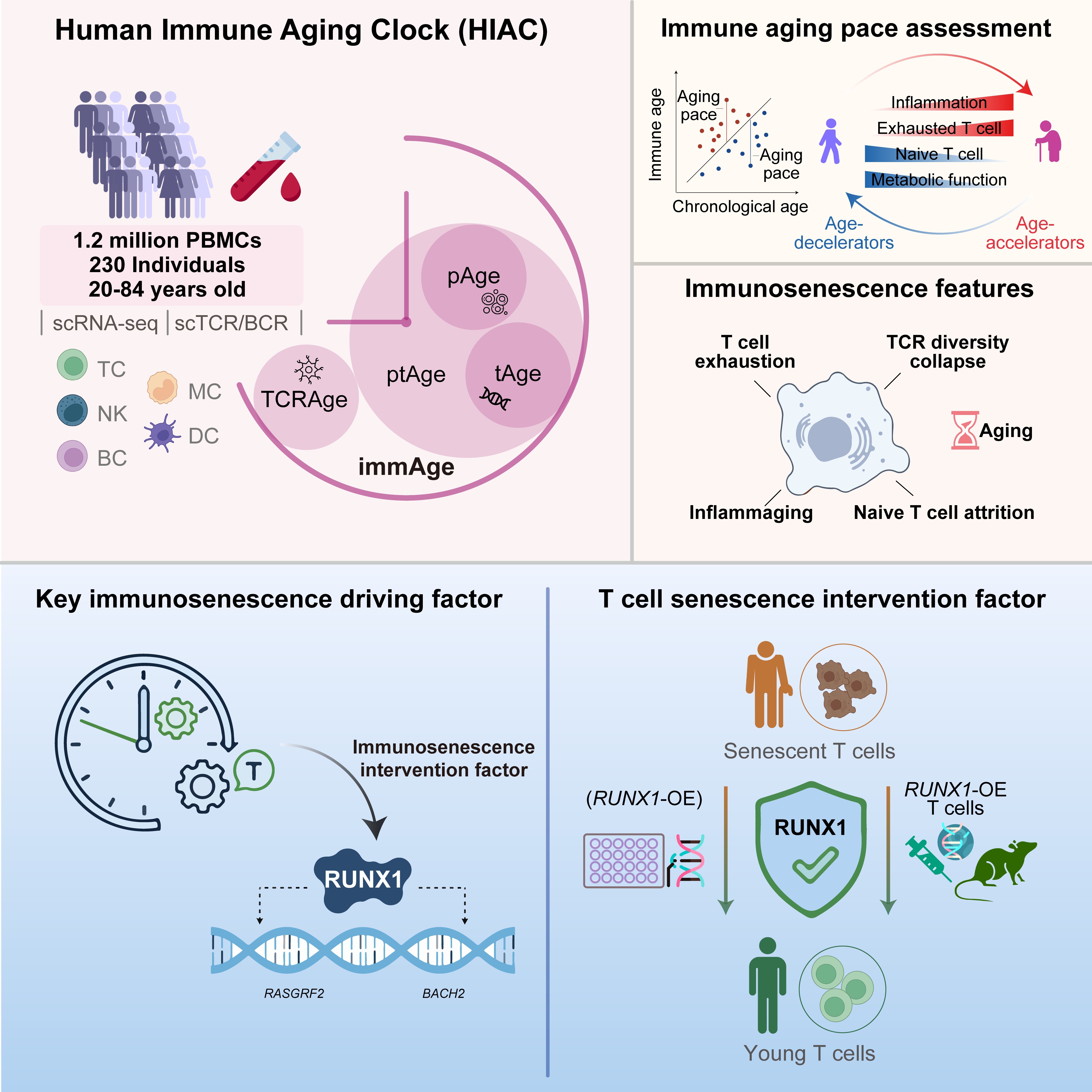
To address these challenges, a collaborative research team led by scientists from the China National Center for Bioinformation, a research center affiliated with the Chinese Academy of Sciences (CAS), together with the CAS Institute of Zoology and Quzhou Affiliated Hospital of Wenzhou Medical University, has constructed a high-precision Human Immune Aging Clock (HIAC). The work systematically characterizes the multiscale dynamics of immunosenescence and identifies the transcription factor RUNX1 as a functional "brake" on T-cell senescence.
The study was published in Immunity on April 14.
To quantitatively assess immune aging, the researchers developed HIAC using single-cell multi-omics data. Traditional approaches to studying immunosenescence have largely relied on single biomarkers or bulk transcriptomic analyses, which fail to capture subtype-specific alterations in immune cells. To overcome this limitation, the team profiled peripheral blood samples from 230 healthy individuals spanning a 60-year age range, generating a high-resolution atlas of nearly 1.2 million peripheral blood mononuclear cells and identifying 24 immune cell subtypes.
The analysis revealed that aging induces profound remodeling of the immune landscape, marked by a sharp decline in naïve T cells and an expansion of exhausted T cells and of monocytes. This pattern reflects a state in which immune exhaustion coincides with chronic inflammation.
Using these data, the researchers established a multilayered immune aging clock framework consisting of pAge (based on cell proportions), tAge (based on cell type-specific transcriptomes), TCRAge (based on T-cell receptor repertoire features), and an integrated multi-modal clock, termed immAge. Among these, immAge achieved a mean absolute error of 5.66 years in predicting chronological age, while T-cell-based clock models performed best among all cell types. These findings demonstrate at single-cell resolution that T cells represent the most sensitive indicators of peripheral immune aging.
Furthermore, the study extended immune aging assessment from population-level patterns to individual-level characterization by introducing the concept of "immune aging pace," defined as the residual of predicted immune age relative to chronological age and derived using linear regression. This metric allowed the researchers to stratify individuals into "age accelerators" and "age decelerators."
Individuals with decelerated immune aging displayed a more youthful immune profile, including higher proportions of naïve T cells, reduced senescence- and inflammation-associated transcriptional signatures, and plasma metabolomic profiles enriched with immunomodulatory and antioxidant molecules. These individuals also exhibited more favorable physiological traits, such as improved glycemic control, better liver function, and enhanced cardiopulmonary capacity.
The results establish a direct link between immune aging dynamics and systemic health, suggesting that immune age may detect early signs of functional decline more sensitively than chronological age.
The study also identified a critical inflection point in human immune aging, with a pronounced peak in cellular and transcriptomic remodeling occurring around age 40. This finding highlights midlife as a key transition period for accelerated immune aging and points to a potential window for preventive intervention.
Through gene regulatory network analysis, the transcription factor RUNX1 emerged as a central regulator, with its downstream target genes contributing strongly to the immune aging clock. RUNX1 acts as a "youth-associated factor" in T cells: maintaining its expression helps counteract senescence, and its overexpression can reverse aging-related phenotypes.
RUNX1 protein levels were found to decline significantly with age in both CD4+ and CD8+ T cells, and its reduction in CD4+ T cells was negatively correlated with the accumulation of the senescence marker γ-H2A.X.
Functional studies confirmed RUNX1 as a key regulator of T-cell senescence. Deletion of RUNX1 in T cells from young donors triggered cell cycle arrest, activation of the senescence-associated secretory phenotype (SASP), telomere shortening in CD8- T cells, and impaired cytotoxic function. Conversely, overexpression of RUNX1 in aged T cells reversed these phenotypes, restoring expression of the co-stimulatory molecule CD27 and lengthening telomeres.
Mechanistically, RUNX1 binds to and activates a network of genes involved in T-cell activation and immune responses, thereby preserving a youthful transcriptional state. In vivo adoptive transfer experiments further supported these results: aged CD8+ T cells overexpressing RUNX1, when transplanted into immunodeficient mice, retained lower levels of senescence markers and higher levels of CD27 expression, demonstrating RUNX1's protective role under physiological conditions.
Collectively, these findings establish RUNX1 not only as a biomarker of T-cell aging but also as a functional regulator, forming a complete evidence chain from association to causation and therapeutic intervention. RUNX1 is thus the first validated intrinsic target for modulating human T-cell senescence.
Human immune aging clock identifies RUNX1 as a decelerator of T cell senescence. (Image by ZHANG Weiqi)
