
The molecular interactions and chemical reactions at metal surfaces are of great importance to many industrial applications. Despite significant progress made in the past decades, it remains extremely challenging to investigate the dissociative chemisorption dynamics of molecular species on surfaces at a full-dimensional quantum mechanical level, in particular for polyatomic-surface reactions.
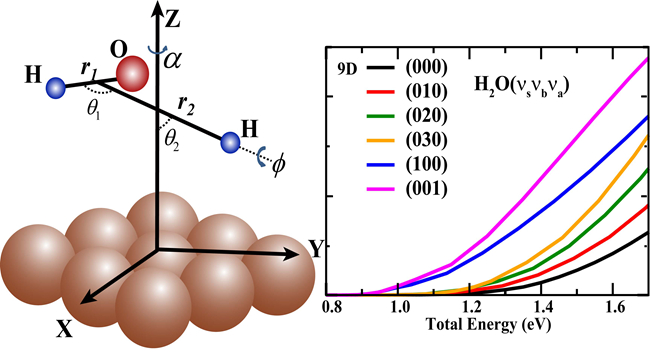
A Dalian Institute of Chemical Physics (DICP) team completed the first full-dimensional quantum dynamics study of a triatomic molecule in the chemisorption process on a rigid metal surface. Specifically H2O/Cu (111) with all nine degrees of freedom (9D) fully coupled on a global potential energy surface and constructed based on extensive DFT calculations, representing a significant step forward in simulating gas-surface reactions from the first-principles.
It is found that the full-dimensional quantum mechanical reactivity is quite different from the corresponding results obtained by previous reduced-dimensional models, indicating the importance of the full-dimensional quantum mechanical calculations to achieve a quantitatively accurate understanding of this reaction. The excitations in vibrational modes of H2O, in particular the stretching modes, are more efficacious than increasing the translational energy in promoting the reaction, much stronger than they were observed in previous reduced-dimensionality quantum studies.
In addition, the characterization of full-dimensional quantum dynamics allows us to exam the validity of two existing approximations regarding the effect of impact sites. Given the validity of the site-averaging approximation confirmed in the H2O/Cu(111) reaction and diatomic molecule-surface reactions, it provides an opportunity to investigate the dissociative chemisorption dynamics of polyatomic molecules on metal surfaces at a full-dimensional quantum mechanical level by site-averaging the reduced-dimensional results over multiple impact sites.
Full-dimensional (9D) and seven-dimensional (7D) site-specific results (Image by FU Bina)
At the beginning of this year, the DICP team reported the first seven-dimensional quantum dynamics study for the dissociative chemisorption of H2O on Cu(111) using the time-dependent wave-packet approach (Chemical Science, 2016, 7, 1840-1845). Very large differences are seen between the 7D dissociation probabilities and the 6D results with fixed azimuthal angles, at different fixed sites of impact. It indicates that the 6D model by neglecting the azimuthal rotation can introduce substantial errors in calculating dissociation probabilities. Hence, a full-dimensional quantum dynamics is essential to investigate the dissociation process.
FU Bina and ZHANG Dong Hui from DICP published their research results in Nature Communications (2016, 7:11953, doi: 10.1038/ncomms11953), entitled “First-principles quantum dynamical theory for the dissociative chemisorption of H2O on rigid Cu(111)”. This is the first time that a full-dimensional quantum dynamics calculation was carried out for the dissociative chemisorption of polyatomic molecules on metal surfaces.
This work was supported by the National Natural Science Foundation of China, the Ministry of Science and Technology of China, and the Chinese Academy of Sciences.

86-10-68597521 (day)
86-10-68597289 (night)

52 Sanlihe Rd., Xicheng District,
Beijing, China (100864)
Copyright © 2002 - Chinese Academy of Sciences











