
Researchers led by Prof. ZHOU Wu from the University of Chinese Academy of Sciences (UCAS) and Prof. Sokrates T. Pantelides of Vanderbilt University have pushed the sensitivity of single-atom vibrational spectroscopy to the chemical-bonding-configuration extreme, which is critical for understanding the correlation of lattice vibrational properties with local atomic configurations in materials.
Using a combination of experimental and theoretical approaches, the researchers demonstrated the effect of chemical-bonding configurations and the atomic mass of impurity atoms on local vibrational properties at the single-atom level.
The study was published in Nature Materials.
In this study, the researchers investigated the atomic vibrations of two types of silicon (Si) point defects in monolayer graphene: the Si-C3 defect, which arises from the substitution of one carbon (C) atom by a Si atom and forms an atomic defect with three bonds with nearest-neighboring C atoms; and the Si-C4 defect, which forms when two C atoms are substituted by one Si atom and results in a defect with four bonds.
The Si-C4 defect produced stronger vibrational signals than the Si-C3 defect in the energy-loss region around 100 meV, suggesting unique vibrational modes for the two defect configurations of the same impurity, according to the researchers.
To examine the extended effect of the two defects, the researchers performed an atom-by-atom analysis focusing on the carbon atoms surrounding the impurities and found that the defects only have a pronounced effect on the nearest neighboring carbon atoms. The next closest neighboring carbon atoms behave almost like typical carbon atoms in graphene.
Remarkably, the researchers found different frequency shifts of the low-energy phonon peak for the nearest neighboring carbon atoms in these two types of Si point defects with different bonding configurations. Complementing this discovery with density-functional-theory calculations, the researchers showed that the different vibrational signals of Si and the nearest neighboring C atoms result from the unique vibrational modes of the two defects, which are primarily dominated by local configurational symmetry.
They also studied another defect with a much lower mass—nitrogen (N) in the form of N-C3. In contrast to Si-C3, the vibrational variation is mainly reflected in the high-frequency peak, which accounts for most of the optical phonon modes. The nearest neighbor extension persists.
This experimental progress was made possible by considerable efforts by the UCAS team to improve the stability of their monochromated scanning transmission electron microscope (STEM) and the sensitivity of the monochromated electron energy-loss spectroscopy (EELS) measurement.
This work has pushed the sensitivity of single-atom vibrational spectroscopy in STEM to the level of chemical bonds and made precise measurements of the vibrational properties of point defects in graphene, providing insights into the defect-induced physics in two-dimensional materials.
This work was supported by the National Key R&D Program of China, the Beijing Outstanding Young Scientists Program, and the National Natural Science Foundation of China.

Figure 1. Vibrational spectroscopy of substitutional Si impurities in graphene with different bonding configurations. (Image by UCAS)
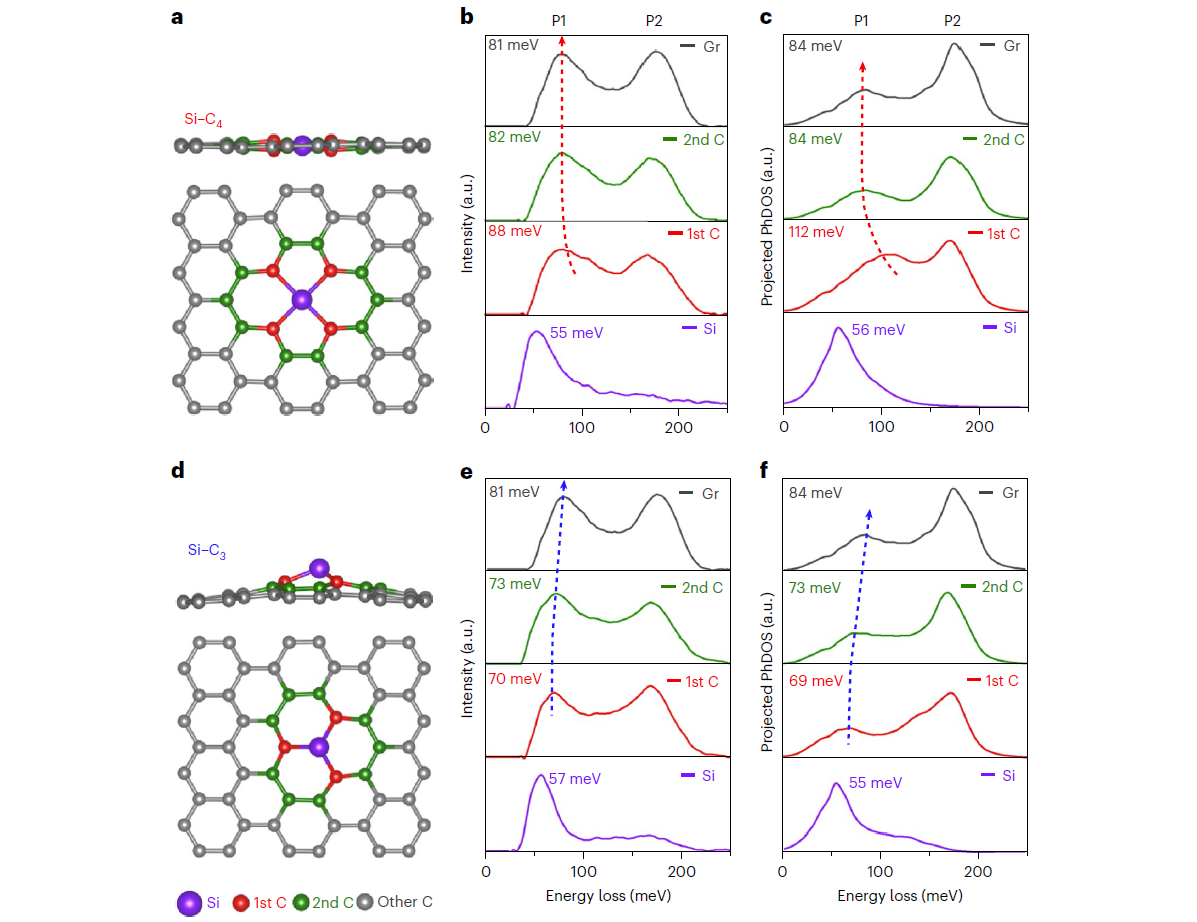
Figure 2. Atom-by-atom analysis of the vibrational EELS spectra in two different types of silicon-point defects in graphene. (Image by UCAS)
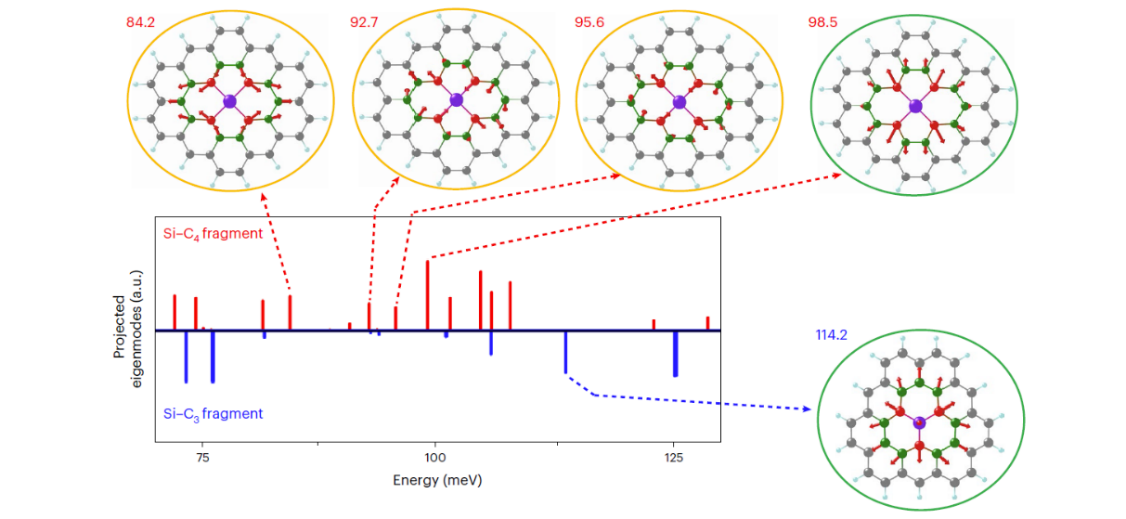
Figure 3. The origin of specific phonon modes observed in Si-C4 and Si-C3. (Image by UCAS)
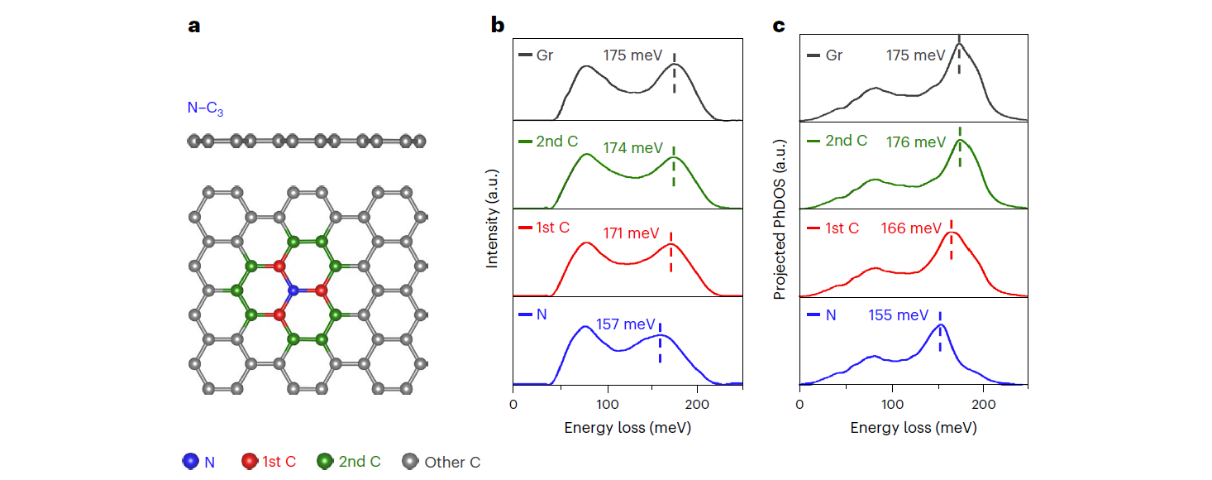
Figure 4. Atom-by-atom vibrational spectroscopy analysis of the N-C3 defect in graphene. (Image by UCAS)
A research group led by WU Zhongshuai from the Dalian Institute of Chemical Physics, in collaboration with WANG Xiaohui from the Institute of Metal Research, developed high-energy-density, hydrogen-ion-rocking-chair MXene based hybrid supercapacitors.

A collaborated team led by Prof. LI Liang and Prof. LI Guanghai at the Hefei Institutes of Physical Science, together with Prof. YAN Feng from Hong Kong Polytechnic University, has recently developed a new two-dimensional-based photodetector with ultrafast photoresponse a...

86-10-68597521 (day)
86-10-68597289 (night)

52 Sanlihe Rd., Xicheng District,
Beijing, China (100864)
Copyright © 2002 - Chinese Academy of Sciences











