
Non-covalent interactions of π faces play a notable role in chemistry and biology. Supramolecular bonding forces involving π faces of carbonyl groups have gained the attention, including lone pair-carbonyl (n…C=O) and carbonyl-carbonyl (C=O…C=O) interactions. These interactions have been shown to control molecular conformations, stabilize peptide/protein structures, regulate reversible covalent systems, and accelerate molecular rotors. The development of synthetic scaffolds exhibiting aromatic-carbonyl interactions in the solid-state and solution hasn't been reported.
In a study published in Advanced Science, the research group led by Prof. YOU Lei from Fujian Institute of Research on the Structure of Matter of the Chinese Academy of Sciences envisaged the proximity of arene plane and aldehyde by using the tetrahedral tertiary sulfonamide serving as a semi-rigid linker to impose restrained conformation, which provides a new platform for studying aromatic-carbonyl interactions.
Researchers prepared a series of ortho-tertiary sulfonamide substituted aromatic aldehydes in a modular way by the reactions of 2-formylbenzenesulfonyl chloride and corresponding amines. X-ray crystal analysis showed that these compounds share common geometric features. Notably, the formyl group was placed above the sulfonamide arene plane, engaging in potential arene-aldehyde interaction. The C…C distances fell within the sum of van der Waals radius and were indicative of attractive interactions. The formyl group rotated in order to interact with aromatic plane on sulfonamide nitrogen.
Conformational analysis of the aldehyde revealed that the most stable rotamer (ON) falls in line with the crystal structure. Two more conformers by virtue of the rotation about S-C and S-N bonds were found, affording relative energies of 0.81 (OFF-1) and 0.78 (OFF-2) kcal/mol, respectively.
Besides, researchers performed Generalized Kohn-Sham energy decomposition analysis to calculate the total interaction energy (ΔETOT) of intramolecular arene-aldehyde/imine interactions in ON conformation and dissect the contributing components. They found that the values of ΔETOT of aldehyde are sensitive to substituent change, with a decreasing sequence (i.e, more negative) from electron-withdrawing NO2 to electron-donating Me2N. Electrostatic (ΔEele) and dispersion (ΔEdisp) terms made a major contribution to ΔETOT, with relative small polarization (ΔEpol) term.
Moreover, researchers quantified the thermodynamic impact of arene-aldehyde/imine interactions on shift imine exchange equilibria and regulated the interactions with solvent effects. Multivariate analysis exhibited a linear correlation of experimental ΔG values versus ΔΔEele, ΔB1, and ΔB5 values. The quality of the correlation was confirmed with an excellent linear relationship between measured and predicted ΔG values, which echoes the electrostatic feature of the interactions.
Researchers then quantified the influence of solvents by correlation with empirical parameters. The -ΔG values of imine exchange reactions in different solvents were plotted against the cohesive energy density (ced) of the solvent, with a linear trendline obtained. Through the competition between imines incorporating secondary and tertiary sulfonamides, researchers found the reversal of kinetic and thermodynamic selectivity and the sorting of dynamic covalent libraries.
In addition, researchers showcased the utility of arene-aldehyde/imine interactions with the diverse modulation of fluorescence signals. Two fluorophores, namely 3-amino-7-diethylaminocoumarin and 4-amino-1,8-naphthalimide, were chosen, and their secondary and tertiary sulfonamides were prepared. Secondary sulfonamides afforded strong blue emission, while tertiary sulfonamides were nearly non-fluorescent. This is likely due to quenching induced by arene-formyl interactions.
Density functional theory (DFT) calculations supported the interpretation, as highest-occupied-molecular-orbital (HOMO) and lowest-unoccupied-molecular orbital (LUMO) orbitals of the tertiary sulfonamides were mainly located on the coumarin plane and the arylaldehyde unit, respectively. Upon the aldehyde reacting with primary amines, the fluorescence was turned on for aliphatic amine derived imines, including 1-butylamine, cyclohexylamine, and t-butylamine, which reveals the increase in the emission with enhanced steric hindrance.
This study lays the foundation for future research on molecular recognition, dynamic assemblies, and smart materials.
Researchers led by Prof. JIANG Ling from the Dalian Institute of Chemical Physics and their collaborators identified a carbon-carbon coupling reaction in neutral transition metal carbonyls.
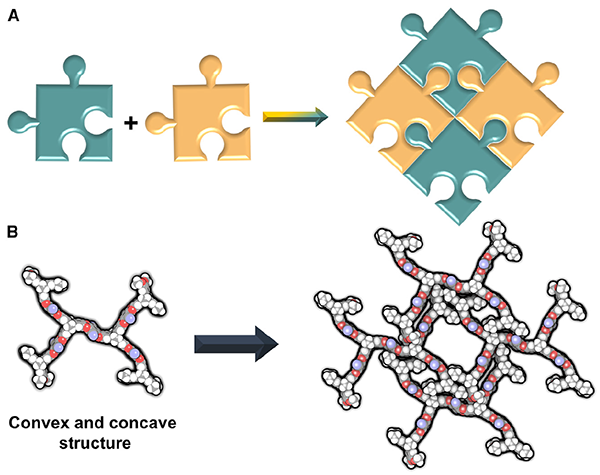
Inspired by jigsaw puzzles, Prof. ZHAO Yanchuan and his research team from the Shanghai Institute of Organic Chemistry have developed a stable, porous molecular crystal through a method they call "interdigitation-directed self-assembly."

86-10-68597521 (day)
86-10-68597289 (night)

52 Sanlihe Rd., Xicheng District,
Beijing, China (100864)
Copyright © 2002 - Chinese Academy of Sciences











