
Newsroom





A single mutation buried within the cell membrane can rewire immune receptor signaling and drive T-cell acute lymphoblastic leukemia (T-ALL), according to a new study published in PNAS on April 28. The study also demonstrates a potential strategy for selectively blocking the disease-causing signaling pathway.
The study was led by Prof. James J. Chou from the Shanghai Institute of Organic Chemistry of the Chinese Academy of Sciences, together with researchers from Zhejiang University in China, Osnabrück University in Germany, and the University of Helsinki in Finland.
Transmembrane domains (TMDs) of cell-surface receptors were previously thought to be structurally passive, serving primarily as membrane anchors. However, growing evidence indicates that these regions play active roles in regulating receptor assembly, conformational coupling, and downstream signaling. Mutations in TMDs of cytokine and immune receptors often cause aberrant, ligand-independent signaling linked to diverse malignancies. However, their underlying mechanisms remain largely unknown.
In this study, the researchers investigated the interleukin-7 receptor (IL-7R), a key regulator of lymphocyte development and homeostasis. They characterized a transmembrane mutation, V253G, which was previously identified in T-ALL, and showed that it drives constitutive signaling in the absence of a ligand. Under normal conditions, IL-7R requires ligand binding and heterodimerization with its co-receptor to activate downstream JAK/STAT signaling. By contrast, the V253G mutation bypasses this requirement, inducing persistent activation of JAK1 and STAT5.
Using an integrative approach combining nuclear magnetic resonance (NMR) spectroscopy, molecular dynamics simulations, biochemical assays, and single-molecule imaging, the researchers demonstrated that the mutation induces an approximately 170° rotation of the TMD dimerization interface, thereby completely altering the receptor's dimerization mode. This structural rearrangement shifts the receptor from a weak, non-activating dimer interface in the wild-type state to a distinct, mutant-specific interface that stabilizes a signaling-competent geometry.
Based on these structural insights, the researchers also rationally designed transmembrane (TM) peptides that selectively block the disease-causing homodimerization. These peptides, delivered via lentiviral infection or lipid nanoparticle (LNP), act as competitive inhibitors to disrupt aberrant homodimerization. Importantly, this strategy effectively suppresses ligand-independent signaling while preserving normal ligand-dependent receptor function.
Together, these findings suggest that understanding how transmembrane mutations reprogram receptor conformation enables the design of targeted TM peptides to selectively block aberrant signaling. These findings provide a conceptual framework for understanding and correcting disease-causing TMD mutations and may be broadly applicable to other cytokine receptors.
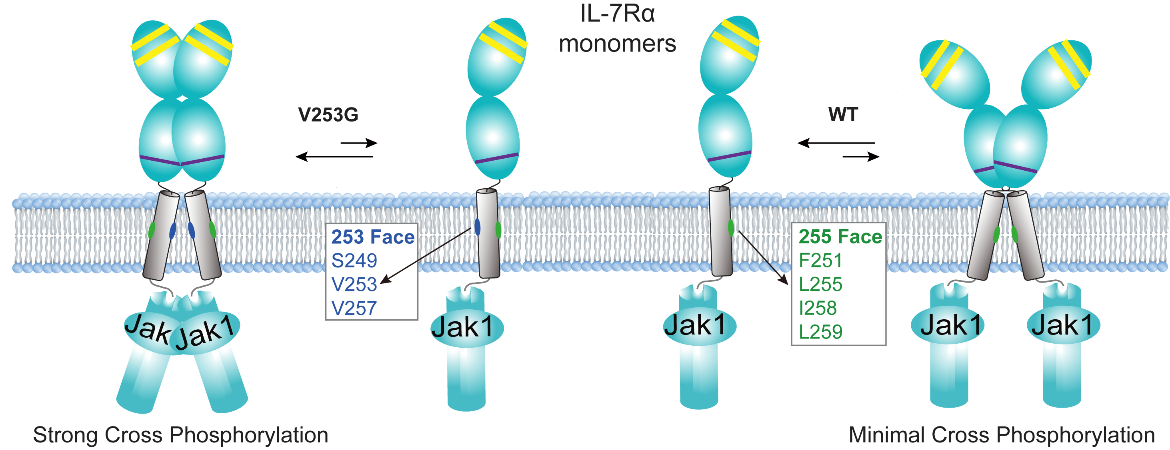
Activating homodimer of V253G IL-7R vs. inactivating homodimer of WT IL-7R. In the V253G IL-7R, residues S249, V253, and V257 form the homodimerization interface, which promotes a JAK1–JAK1 pairing configuration that leads to strong cross phosphorylation. In contrast, the WT IL-7R forms a homodimerization interface composed of F251, L255, I258, and L259, which is incompatible with JAK1-JAK1 cross phosphorylation. (Image by James J. Chou)
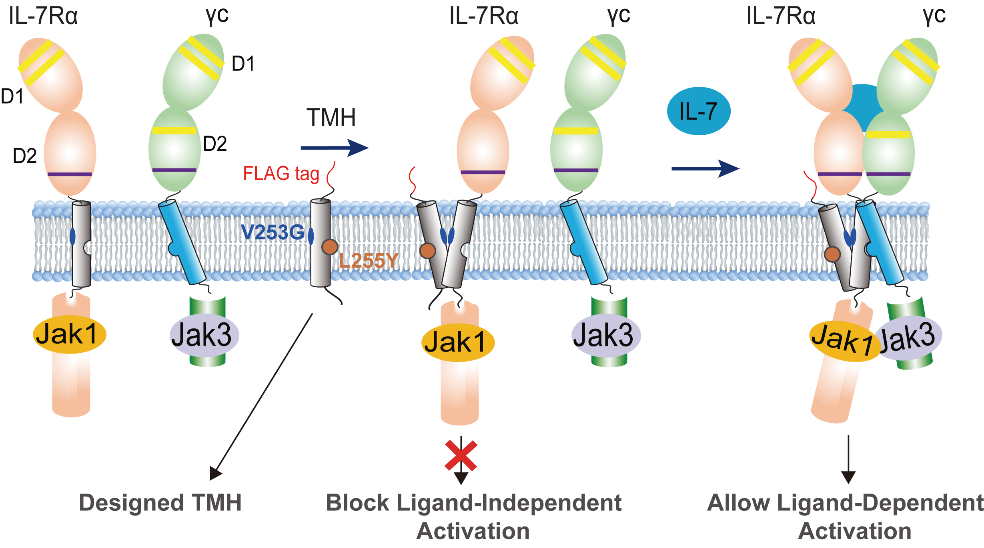
Schematic illustration of the TMD design concept for competitive blocking of IL-7R V253G homodimerization and ligand-independent receptor activation but not affecting IL-7 induced signaling. (Image by James J. Chou)


