
Newsroom





Heterogeneous catalysis—in which catalysts and reactants are of different phases, e.g., solid and gas—is important to many industrial processes and often involves solid metal as the catalyst. Ammonia synthesis, catalytic converters for automobile exhaust, methanol synthesis, carbon dioxide reduction, and hydrogen production are examples of such metal-catalyzed heterogeneous catalysis.
The electronic structure of metal surfaces governs the adsorption of reactants and intermediates, and thus the catalytic activity. For this reason, strain engineering—which tunes the electronic structure of a metal catalyst by stretching or compressing its crystal lattice—has emerged as an important strategy for enhancing catalytic performance. Unfortunately, scientists have not been able to quantify how metal strain influences adsorption energies and reaction barriers across different metal catalysts, thereby limiting the rational design of catalysts with desired properties.
To address this challenge, a research team from the Lanzhou Institute of Chemical Physics (LICP) of the Chinese Academy of Sciences has developed a method to predict how strain modifies adsorption energies and reaction barriers across diverse metal systems.
The team's findings were published in Cell Reports Physical Science on March 31.
Through systematic density functional theory calculations on multiple transition metal surfaces, the researchers found that the energetic response of an adsorbate to lattice strain is closely correlated with its electronegativity. This insight allowed the researchers to classify adsorbates into two groups: those with low electronegativity and those with high electronegativity. The scientists used how carbon–hydrogen (CH) and oxygen (O) respond to strain on the metal surface as reference points to classify these groups.
Based on this classification, the team derived the dual-descriptor linear scaling relationship ΔE(ads) = a·ΔE(O) + b·ΔE(CH) + c. The model outperforms conventional single-descriptor methods and extends successfully to reaction transition states, enabling the quantitative description of energy barrier changes under strain.
To explore the physical origin of this phenomenon, the team adopted machine learning to build a classifier using Bader charge transfer and the integrated crystal orbital bond index (ICOBI) as key descriptors. The results revealed that differences in the degree of covalent versus ionic bonding between adsorbates and the metal surface are the fundamental reasons why strain responses follow an electronegativity-dependent pattern across different adsorbates.
This work provides a unified, transferable framework for the rational design of strain-engineered catalysts, supporting the design of strain-engineered catalysts for key reactions including CO2 conversion, nitrogen reduction, and hydrogen evolution.
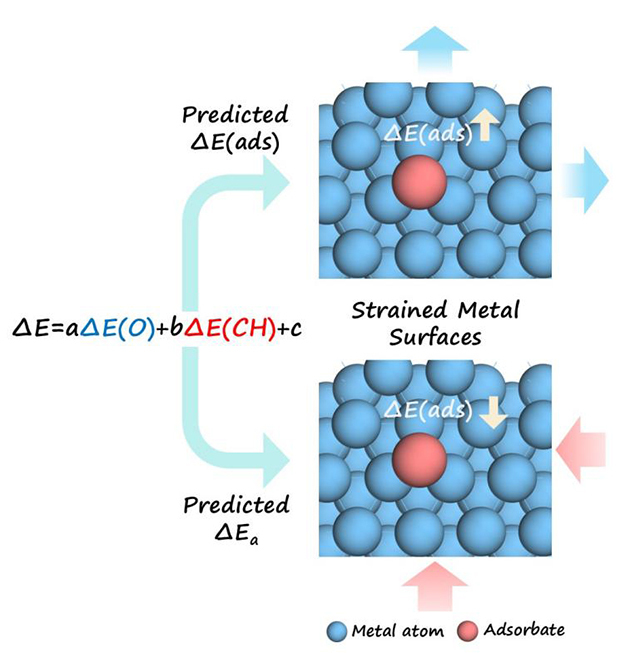
Schematics for the dual-descriptor scaling relation. (Image by LICP)
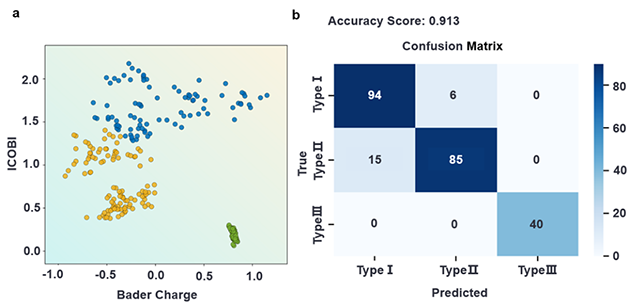
Classification of the different characteristics of strain effects using ICOBI and Bader charge. (Image by LICP)
